Events
Genetic characteristics of highly pathogenic avian influenza (HPAI)
Release date : December 15, 2016 (Thursday)
Overview
- In the National Institute of Animal Health NARO, we identified the subtypes of highly pathogenic avian influenza (HPAI) virus which isolated from Aomori and Niigata cases on November 28. We performed the whole genome analysis to estimate the origin and pathogenicity of the virus at the gene level.
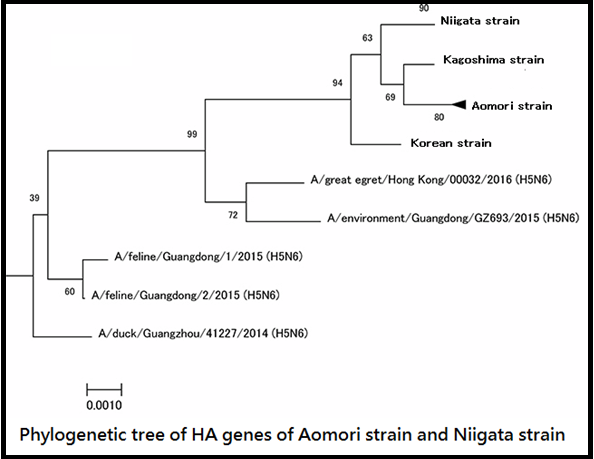
- We identified HPAI causative virus (Aomori strain, Niigata strain) which isolated in Aomori and Niigata Prefectures as H5N6 subtype highly pathogenic avian influenza virus (HPAIV). Using the next-generation sequencer, we determined the nucleotide sequences of 99.5% (Aomori strain) and 100% (Niigata strain) of each virus genome. The
nucleotide sequences of the two viruses have mutual homology of 99.2% or more in all of the 8 RNA segments each other. In addition, both viruses showed 99.3% or more homology with the virus which is isolated from the water of the nesting place of hooded crane of Izumi City, Kagoshima.
- When comparing the 8 RNA segments with influenza virus genes which is published in the public gene database, it was revealed that both Aomori and Niigata strains have homology of 97.4% or more with the H5N6 subtype HPAIV (Korean strain) isolated in Korea in all segments. In addition, seven gene segments out of the eight gene segments and the remaining one segment showed more than 97% homology with the H5N6 subtype HPAIV that was in circulation in 2015 or the other avian influenza virus (AIV), respectively.
※ However, this result does not indicate the origin of the virus directly.
- Based on the amino acid sequence deduced from the determined nucleotide sequence, we assessed the risk of human infection. Since strains from Aomori and Niigata doesn't have amino acid mutation which is thought to be involved in human infectivity, it is considered that the possibility is low that this virus infect human directly.