The National Institute of Animal Health, NARO (NIAH) performed genetic sequence analysis of the causative virus of classical swine fever which was isolated from an outbreak in Gifu Prefecture, Japan for the first time in 26 years.
The classical swine fever virus is divided into three groups (genotypes) and is further subdivided into subgroups (subgenotypes) according to the nucleotide sequence of a specific region of the viral gene. The virus isolated from infected pigs in Gifu Prefecture on September 9, 2018 belongs to the subgenotype 2.1 group based on the sequence of the classical swine fever viruses registered in the international database of nucleotide sequences.
Many vaccine strains, including the vaccine strain previously used in Japan belong to the genotype 1 group. In addition, viruses that belong to subgenotype 2.1 have not been detected in Japan so far. However, the viruses belonging to subgenotype 2.1 have been epidemic already in Europe and Asia, hence the causative virus intruded Japan from overseas. Detailed analysis of antigenicity and pathogenicity including experimental infections will be carried out in the future.
Reference Information
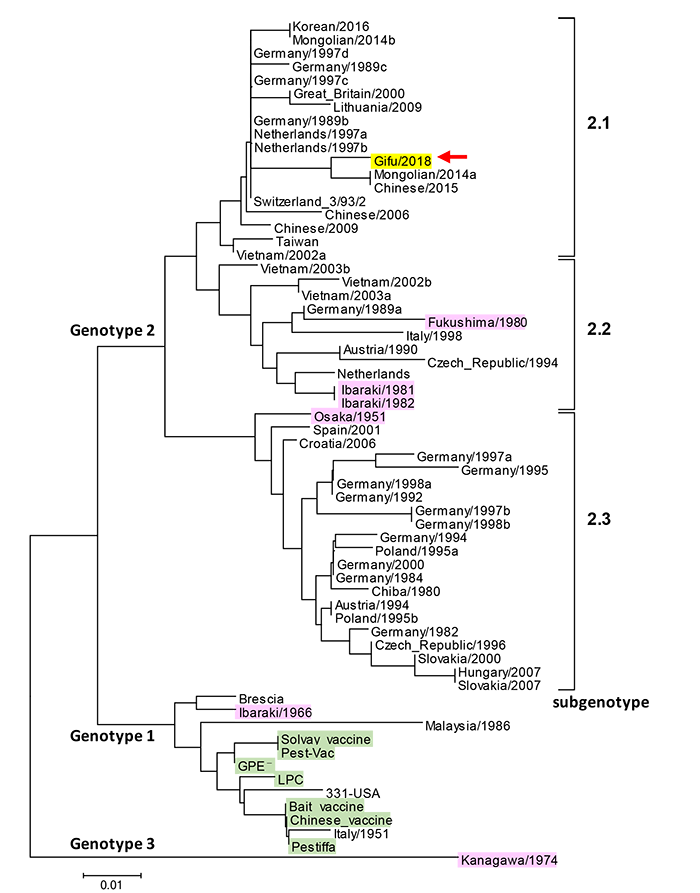
Phylogenetic tree of the classical swine fever virus gene
(Yellow: Gifu strain, Pink: Strain of domestic virus detected in the past, Green: Vaccine strain)
The genotype reported by Postel et al. (Veterinary Research 2012, 43:50) was used as reference. Analysis of the domestic strain and recent Asian epidemic strains was done by the neighbor joining method using a 150 bp of 5'untranslated region (Analysis software: Mega 7.0 Software. Alignment method: Clustal W ).






